Risk
Risk thinking must stay connected
Teams need better linkage between quality events, change, and documented risk decisions.
Medical device manufacturers need ISO 13485–aligned QMS execution with ISO 14971 risk thinking visible in design changes, CAPA, and documentation — so training, records, and release decisions stay inspection-ready for notified bodies and FDA. The compliance hub stays centered on validation, data integrity, Part 11 records, and security — the evidence set device QA teams defend in audits.

The quality and compliance pressures unique to your regulated market — and how Complere is built to address them.
Teams need better linkage between quality events, change, and documented risk decisions.
Procedural updates and qualification gaps can quickly become inspection questions.
Backlogs, recurring issues, and design-impacting changes cannot remain siloed.
The regulatory frameworks, guidance documents, and audit expectations that govern quality operations in this sector.
The international QMS standard for medical device manufacturers — covering document control, CAPA, training, internal audits, and supplier qualification throughout the product lifecycle.
Risk management for medical devices — requiring documented risk identification, analysis, evaluation, mitigation, and residual risk acceptance at each stage of product design and change.
The FDA Quality Management System Regulation — the US quality system requirements for device manufacturers, updated in 2024 to align with ISO 13485 and harmonise global device QMS expectations.
The EU Medical Device Regulation — the mandatory framework for CE marking, post-market surveillance, clinical evaluation, and quality system requirements for devices sold in European markets.
The FDA electronic records and electronic signatures regulation — governing audit trail requirements, access controls, signature meaning statements, and record integrity for digital quality systems.
The EU GMP computerized systems guideline — covering validation, audit trails, data integrity, access controls, and backup requirements for quality software used in regulated device manufacturing.
The Complere modules most commonly used by teams in this industry to manage compliance and quality operations.
Maintain design history files, device master records, and controlled SOPs under version-controlled lifecycle governance — meeting ISO 13485 document control requirements and FDA QMSR record accessibility expectations.
Learn moreInvestigate nonconformities with structured root cause analysis, link findings to ISO 14971 risk records, and track corrective actions through effectiveness review for ISO 13485 clause 8.5 compliance.
Learn moreAssess design and process changes against EU MDR traceability requirements and QMSR impact criteria — with linked documentation, validation evidence, and approval sign-off captured in one traceable record.
Learn moreAggregate CAPA trends, audit findings, complaint data, and risk signals into ISO 13485–structured management review inputs — giving leadership the cross-module visibility required for clause 5.6 periodic review.
Learn moreExplore related modules, compliance topics, and guides to build a complete picture of your quality system.

Explore this topic in more depth to build a complete picture of your quality and compliance operations.
Explore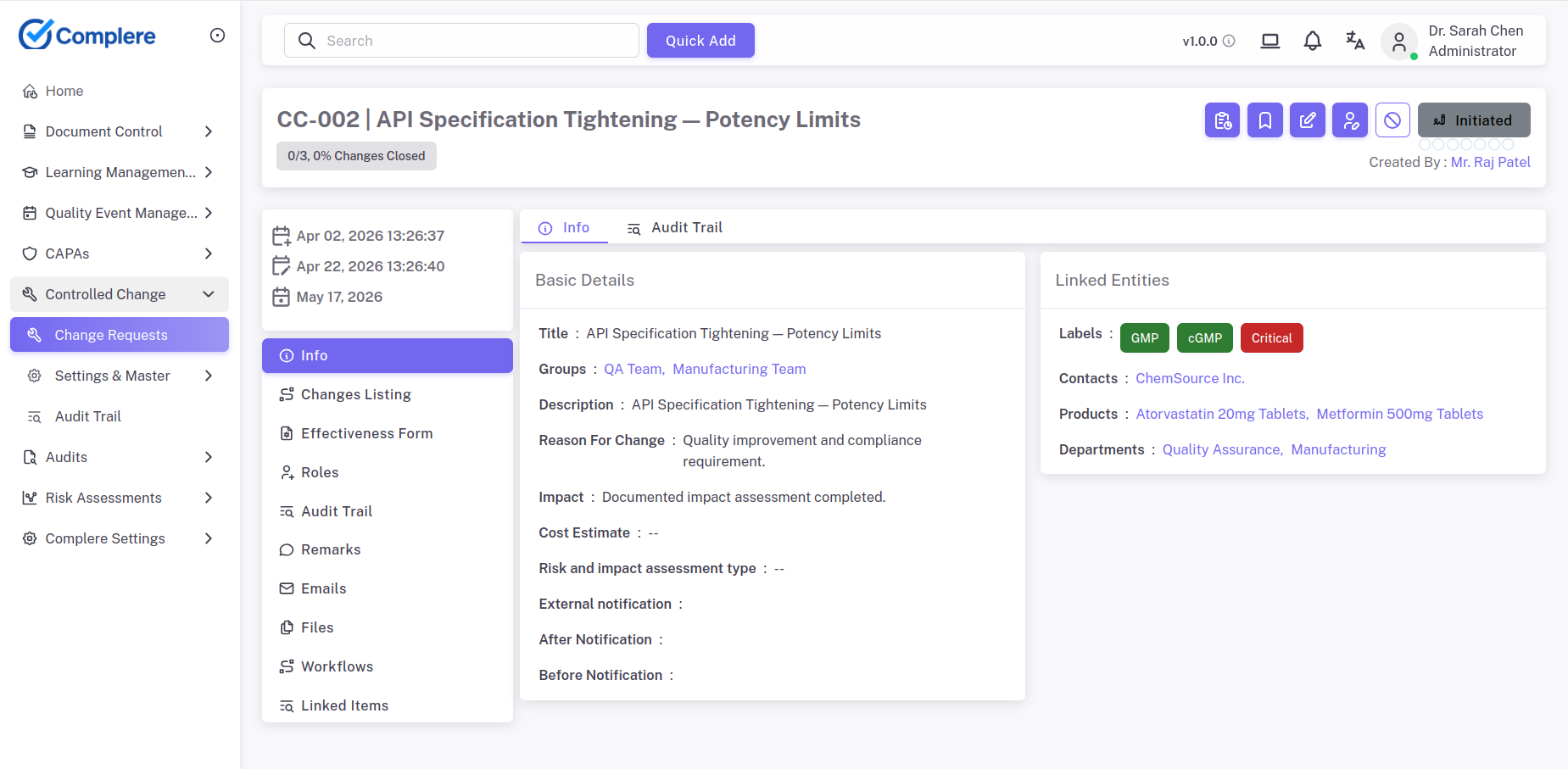
Explore this topic in more depth to build a complete picture of your quality and compliance operations.
Explore
Explore this topic in more depth to build a complete picture of your quality and compliance operations.
ExploreOur demos cover device-specific quality workflows — risk management, device history records, CAPA, and training competency — in your context.